





")

")











The signs of Prader-Willi syndrome can vary significantly with age, often leading to early misdiagnosis. A newborn with Prader-Willi syndrome may be floppy, weak, and unable to feed well, while an older child may develop an intense, constant drive to eat. These very different presentations are part of the same condition, caused by the same genetic problem.
Understanding how symptoms change across the lifespan is important because early diagnosis leads to better outcomes. Timely treatment, particularly growth hormone therapy and structured nutrition, can significantly reduce many complications. PWS affects about 1 in 15,000–25,000 births and occurs equally in populations.
Read More: Genetic Testing for Hypertrophic Cardiomyopathy: Should Your Family Get Screened?
- Prader–Willi syndrome is a genetic condition caused by loss of function of paternal genes on chromosome 15, and its symptoms change significantly with age.
- It begins with low muscle tone and feeding difficulties in infancy, then shifts to hyperphagia, developmental delays, and distinctive behavioral patterns in childhood.
- Early diagnosis and lifelong management, including growth hormone therapy and strict nutritional control, are essential to reduce complications and improve quality of life.
What Causes Prader-Willi Syndrome
Prader-Willi syndrome is caused by the loss of function of genes on a specific region of chromosome 15 (15q11.2–q13), specifically the copy inherited from the father.
There are three main genetic mechanisms:
- About 70% of cases are due to a deletion in the father’s chromosome 15
- About 25% occur when the child inherits two copies of chromosome 15 from the mother and none from the father (maternal uniparental disomy, or UPD)
- About 5% are due to an imprinting defect, where paternal genes are present but not active
All three lead to the same outcome: important genes that regulate hunger, hormones, growth, and behavior are not expressed.
This same chromosomal region is involved in Angelman syndrome, but that condition affects the maternal copy instead, which is why the two are sometimes considered during early genetic evaluation.
Signs in Newborns and Infants
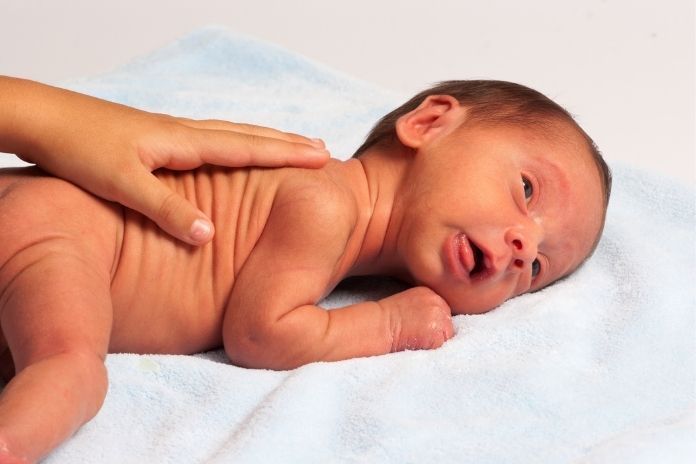
Low muscle tone and feeding problems dominate the first phase of Prader-Willi syndrome. These early signs are often subtle but highly characteristic.
Hypotonia: Hypotonia (low muscle tone) is the most consistent feature and is present in nearly all infants with PWS. Babies feel noticeably floppy when held, with little resistance in their limbs. Their movements are reduced, and their cry is often weak.
Feeding Difficulties: Feeding difficulties are common due to a “poor suck reflex” and low muscle tone. Many infants cannot feed effectively and may require nasogastric tube feeding. Despite reduced intake, weight gain is poor, often leading to a diagnosis of failure to thrive.
Physical Features Visible at Birth: Physical features may be present at birth, though they can be mild. These include almond-shaped eyes, a narrow forehead (bifrontal diameter), and a downturned mouth. You may also observe hypopigmentation, lighter skin, and hair compared to family members. In boys, undescended testicles are common.
Lower Fetal Movement: Reduced fetal movement is sometimes reported during pregnancy, which may be an early clue but is not always recognized at the time.
Signs in Early Childhood — The Phase Transition
Between early childhood and school age, the condition shifts from poor feeding to excessive hunger. This is a key transition in Prader-Willi syndrome.
When Hyperphagia Begins
Between ages 1 and 6, children typically develop hyperphagia, which is an intense and persistent drive to eat.
This is not typical hunger. Children with PWS do not respond normally to satiety signals; they do not feel full after eating. As a result, they may eat continuously, seek food constantly, and become preoccupied with it in a way that resembles obsessive-compulsive behavior.
Without strict supervision, this behavior leads to rapid weight gain and a high risk of severe obesity. This phase reflects dysfunction in the hypothalamus, the part of the brain that regulates hunger and fullness.
Developmental And Physical Signs In This Period
During this stage, developmental delays become more noticeable. Motor milestones such as walking are often delayed; also, coordination is affected.
Most children also experience delays in speech and language development. At the same time, short stature becomes more apparent due to growth hormone deficiency, which is present in most individuals with PWS.
Other common physical features include small hands and feet, strabismus (crossed eyes), and scoliosis, which requires monitoring.
As hyperphagia leads to weight gain, additional health risks begin to appear, including type 2 diabetes and sleep apnea.
Read More: Insulin Resistance in Children and Adolescents: What Parents Should Know
Behavioral Signs — A Distinctive Profile

The behavioral features of Prader-Willi syndrome are core to the condition and reflect underlying brain differences.
Common patterns include frequent tantrums, especially when food access is restricted or routines are disrupted. Individuals often show rigidity, meaning they struggle with changes and prefer strict routines and predictability.
Obsessive-compulsive traits are also common. These may include repetitive questioning, hoarding, and a strong focus on specific topics, particularly food.
Skin-picking (excoriation) is a well-known behavior and can lead to noticeable skin damage. Another important feature is a high pain threshold, meaning injuries may go unnoticed or unreported.
In some adolescents and adults, particularly those with maternal uniparental disomy, psychiatric symptoms such as psychosis can occur.
These behaviors are not simply intentional; they are part of the neurological profile of the condition.
Signs in Adolescents and Adults
As individuals with PWS grow older, hormonal and metabolic issues become more prominent.
Hypogonadism is present in both males and females and leads to incomplete puberty. Girls may have minimal breast development and irregular or absent menstrual cycles, while boys may have limited testicular development and reduced facial hair. Most individuals are infertile.
Bone density is often reduced due to the combined effects of growth hormone deficiency and hypogonadism, increasing the risk of fractures over time.
Food-related challenges continue throughout life. Most individuals require strict environmental controls, such as supervised meals and restricted access to food, to prevent obesity.
Without these measures, obesity-related complications, including cardiovascular disease, type 2 diabetes, and sleep apnea, are the main health risks.
Intellectual disability is present in all individuals with PWS and ranges from mild to moderate. With structured support, many adults can work and live in supervised or semi-independent settings.
With proper management, life expectancy is not significantly reduced, but complications related to obesity remain a leading cause of serious health issues.
Diagnosis and Treatment — What Happens After Suspicion

Diagnosis
When Prader-Willi syndrome is suspected, especially in a newborn with hypotonia, genetic testing should be performed.
DNA methylation analysis of the chromosome 15q11.2–q13 region detects more than 99% of cases and is the recommended first-line test. It confirms the diagnosis but does not identify the exact genetic mechanism.
Further testing, such as chromosomal microarray or UPD analysis, is used to determine the underlying cause, which is important for genetic counseling.
Treatment
There is no cure for Prader-Willi syndrome, but management is multidisciplinary and lifelong.
Growth hormone therapy, often started in infancy, improves height, muscle tone, body composition, and bone density. Because growth hormone deficiency is present in most individuals, this treatment plays a central role.
Strict control of food access is essential and should begin early, before hyperphagia becomes established. Structured routines and environmental controls are key to preventing obesity.
Additional treatments include speech therapy, physical therapy, behavioral interventions, and hormone replacement therapy during adolescence to support puberty and bone health.
In March 2025, a new option became available: VYKAT XR (diazoxide choline extended-release), the first medication approved specifically to treat hyperphagia in individuals with PWS, aged 4 and older.
Read More: How Muscle Mass Affects Longevity — and How to Maintain It After 40
Conclusion
Prader-Willi syndrome is a condition that changes significantly over time. The condition progresses from a floppy newborn with feeding difficulties to a child with hyperphagia and subsequently to an adult managing lifelong hormonal, behavioral, and metabolic challenges.
Recognizing early signs, especially hypotonia and feeding problems in infancy, is critical for timely diagnosis. Early intervention, including growth hormone therapy, dietary management, and supportive care, can greatly improve outcomes and quality of life. If these signs are present, referral for genetic evaluation is an important next step.
In this Article


